When we are working on individual genes or proteins, or we just want to take a closer look at sequences, the most intuitive method is to use an alignment viewer.
There are a number of different alignment viewers available such as Jalview and NCBI’s own (MSA) – you can find a list of many more here .
Popular software such as Geneious and MEGA also have their own in-built alignment viewers, though pre-warning Geneious isn’t free!
For this post we’ll explore Aliview, a free and intuitive alignment viewer available here.
It’s lightweight and comes with some handy inbuilt tools to help visualise and edit sequences, let’s try it out with an example!
Example: Exploring a COX-1 alignment in Aliview
In this example we will use an alignment of COX-1 coding sequences from parasite species in the genus, Schistosoma.
Visualising a sequence alignment
Taking a look at the alignment below, we notice Aliview, like many other alignment viewers, assigns a colour to each nucleotide (A,C,G,T).
This makes it easier to spot the similarities and differences in the alignment by eye.

For example, we can see conserved blue Cytosine nucleotides.
These nucleotides are shared by all species in the alignment, most notably at positions 39, 40, 75 and 76.
Conversely, there are positions in the alignment which have high levels of variation, for example at position 80, where we have multiple SNPs.
Highlighting nucleotides that differ from a selected sequence
This useful functionality highlights the nucleotides which differ specifically to a chosen sequence.
In this example I have selected the Schistosoma mansoni sequence.
To highlight your chosen sequence in Aliview you can simply click on the sequence before selecting the option illustrated below.
Using this method we can see where, along the sequence, some nucleotides differ in other species compared to Schistosoma mansoni.

Highlighting nucleotides that differ from the consensus sequence
Another useful functionality in Aliview is highlighting the difference from a consensus sequence.
The consensus sequence is a sequence representing the alignment which is made from the most frequently occurring nucleotides at each position (think of it as a summary of the sequence data).
By highlighting the less frequently occurring nucleotides in this way we can quickly identify any variable sites or regions along the alignment.
This option can be selected as shown below:

Protein translation
In Translating open reading frames (orfs) we ran through using the program Transeq to translate a DNA sequence into it’s corresponding Amino Acid sequence.
In Aliview and some other alignment viewers, there is functionality to perform translation and then find the longest open reading frame visually.
As with nucleotide sequences, the different Amino acids are assigned a colour to help with interpretation.
To translate the sequence we first select the option ‘Translates nucleotide sequence to Amino acids‘ illustrated by arrow 1 below, then we select ‘Show translation as only one character Amino acid‘ illustrated by arrow 2:

Now we have our single character Amino acid sequence!
In the top right of the viewer we can see 1.Standard code with a drop down menu, this means our alignment has been translated by default using the standard translation table.

You may notice that the alignment contains X’s at some positions, which are not assigned a colour.
So why are these X’s there?
If a sequence contains N’s instead of A,C,T or G in a codon, the amino acid will be assigned as X during translation.
X’s can also show up when we have in-frame stop codons and unless we are expecting to see them (for example, if we have selenocysteine) they indicate our sequence is out of frame.
This can be caused by using the wrong translation table and/or choosing the wrong reading frame.
Let’s change it to the correct translation table for these COX-1 sequences, which is Invertebrate mitochondrial:
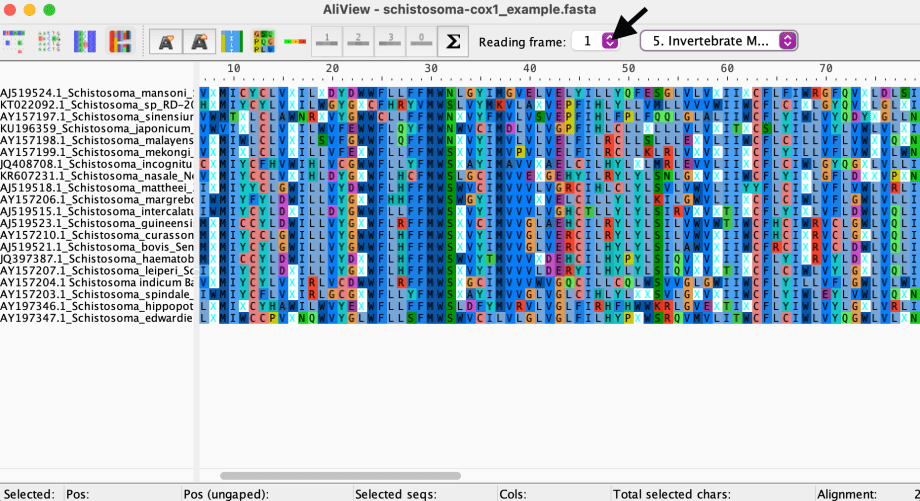
Look’s like we still have some in-frame stop codons ( X‘s ). This means we need to try the other reading frames.**
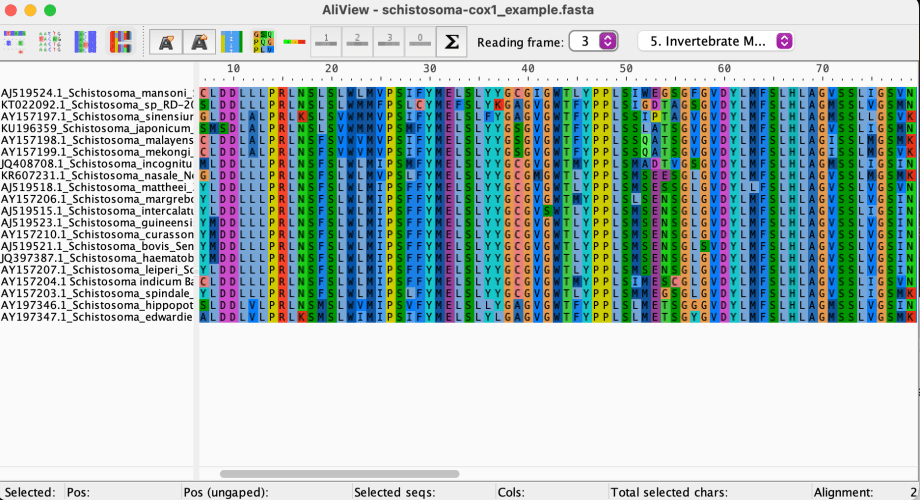
To do this, select the drop down menu next to Reading Frame: and try frames 2 and 3.
Bingo! No more X’s, we can see that frame 3 is the longest ORF for these sequences.
Selecting sequence regions
In many alignment viewers you can manually edit the sequence text.
This is useful if you want to trim the alignment to the same length or you wish to extract parts of a sequence to save in another fasta file.
To select sequence regions you can simply click and drag across the viewer.
For example, for trimming sequence ends we can manually select the sequence region by dragging our mouse over the region we wish to delete, then right click and select ‘Delete selected sequence(s)‘ as shown below:

Reformatting alignments
Aliview also lets you save an alignment in a number of different formats. This is necessary when using bioinformatic programs which require a specific format.
To save the alignment in a different format go to File and find the appropriate Save as option:

There are many other functionalities available in Aliview such as merging two sequences, deleting gaps and performing re-alignment.
In an era of genomics and big data we won’t always want to do manual editing however once you have an eye for sequence data, visual inspection becomes invaluable when you need to inspect sequence regions in detail.
I hope you enjoyed this quick introduction to using an alignment viewer!